导语
前沿科研成果
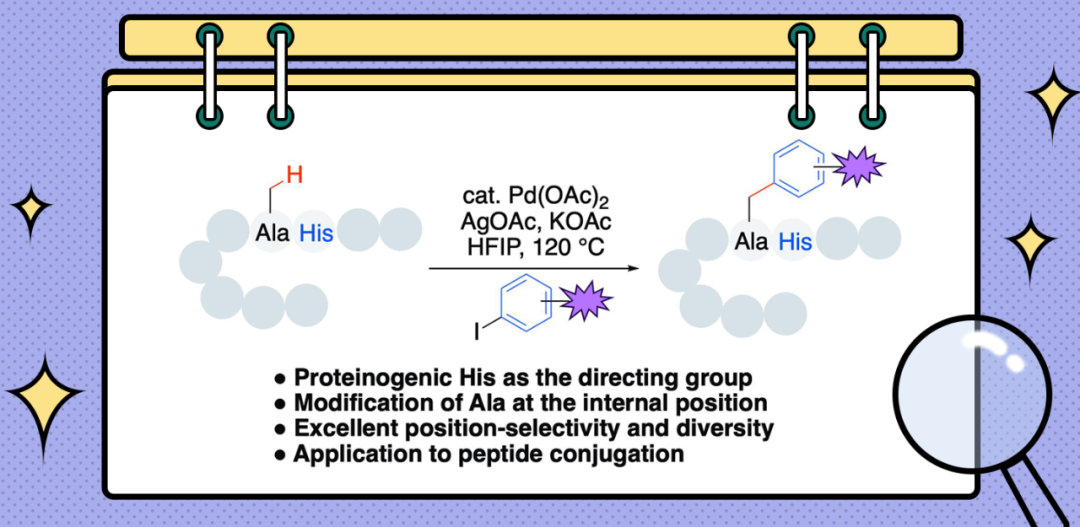
基于组氨酸导向非末端丙氨酸β-C(sp3)-H芳基化方法的多肽组装后修饰
Ackermann和翁意意首次使用含配位性侧链的氨基酸作为内源性导向基团,用于多肽侧链sp3碳氢键官能团化。他们先后发展了天冬酰胺和天冬氨酸[3]导向的β-C(sp3)-H官能团化方法,实现了多肽氮端丙氨酸残基的β-C(sp3)-H芳基化以及炔基化。之后,陈弓[4]和姚波[5]几乎同时报道了蛋氨酸导向的C(sp3)-H官能团化方法,分别用于线性肽氮端氨基酸残基的γ-/β-C(sp3)-H芳基化和首尾连接环肽丙氨酸残基的β-C(sp3)-H芳基化。尽管取得了以上重要进展,这些方法用于线性肽的非末端丙氨酸残基的β-C(sp3)-H芳基化时遇到较大的困难,主要原因可能是多肽骨架酰胺键与钯催化剂的配位形成非活性中间体而表现出的抑制效应。
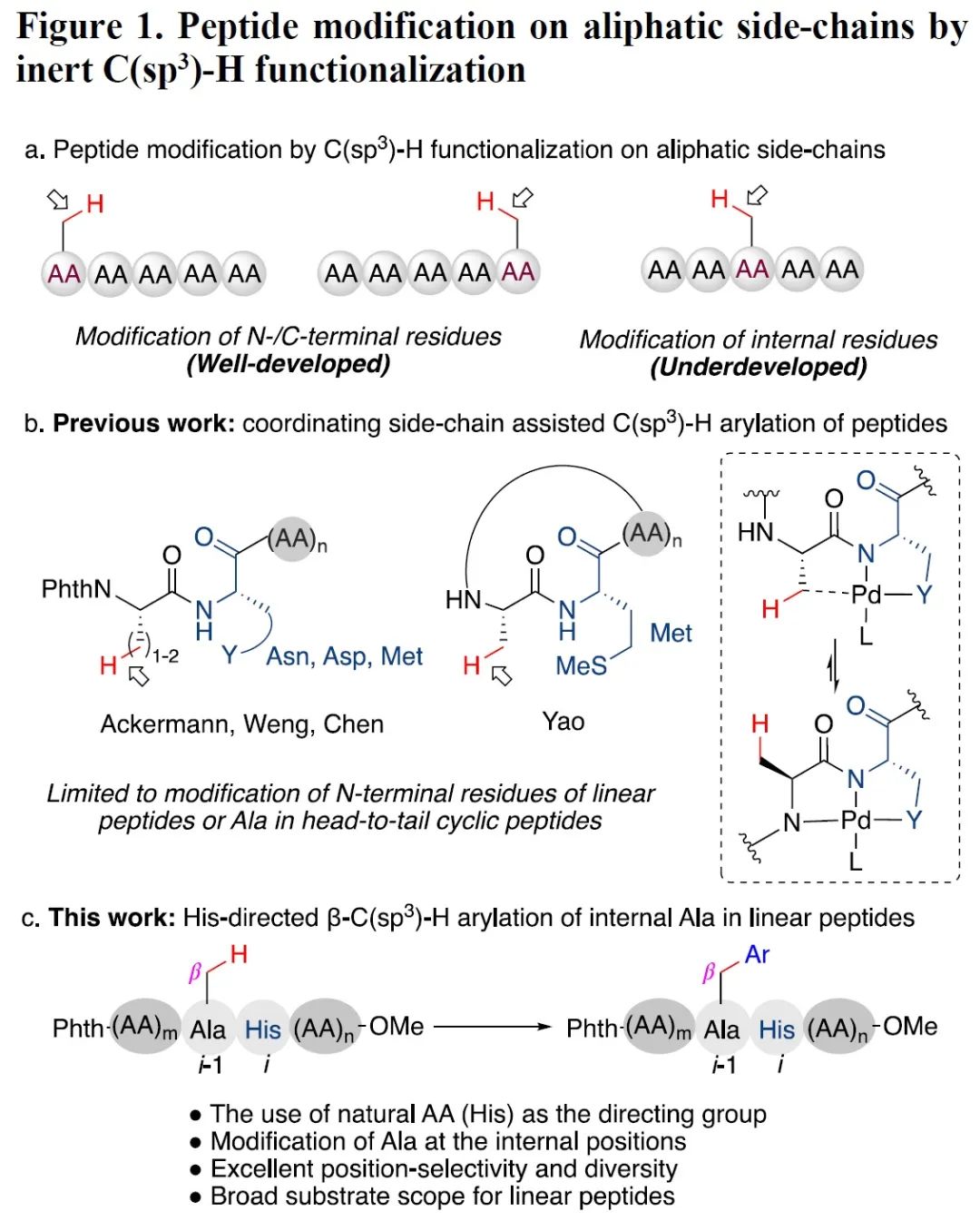
L-组氨酸存在于许多天然多肽和蛋白质的活性区域,在催化反应、金属离子传输等过程发挥关键作用。基于二价铜离子和镍离子与组氨酸残基的优良结合能力,Ball[6]发展了铜/镍催化多肽和蛋白质的骨架酰胺N-H键的Chan-Lam芳基化/烯基化修饰。受此报道的启发,我们设想以二价钯作为催化剂,利用钯与组氨酸类似的配位能力,同时考虑到二价钯具有相对二价铜和镍较软的性质,削弱骨架酰胺键对钯的配位作用,减轻骨架酰胺键的抑制效应,最终实现钯催化组氨酸导向的碳氢键官能团化。因此,本论文报道了一种组氨酸导向多肽侧链的β-C(sp3)-H芳基化方法。该工作的主要亮点如下:1)首次利用蛋白源组氨酸残基作为导向基团用于碳氢键官能团化;2)针对多肽非末端丙氨酸残基的选择性修饰;3)反应表现出优良的位置选择性和多样性;4)底物适用范围较广。
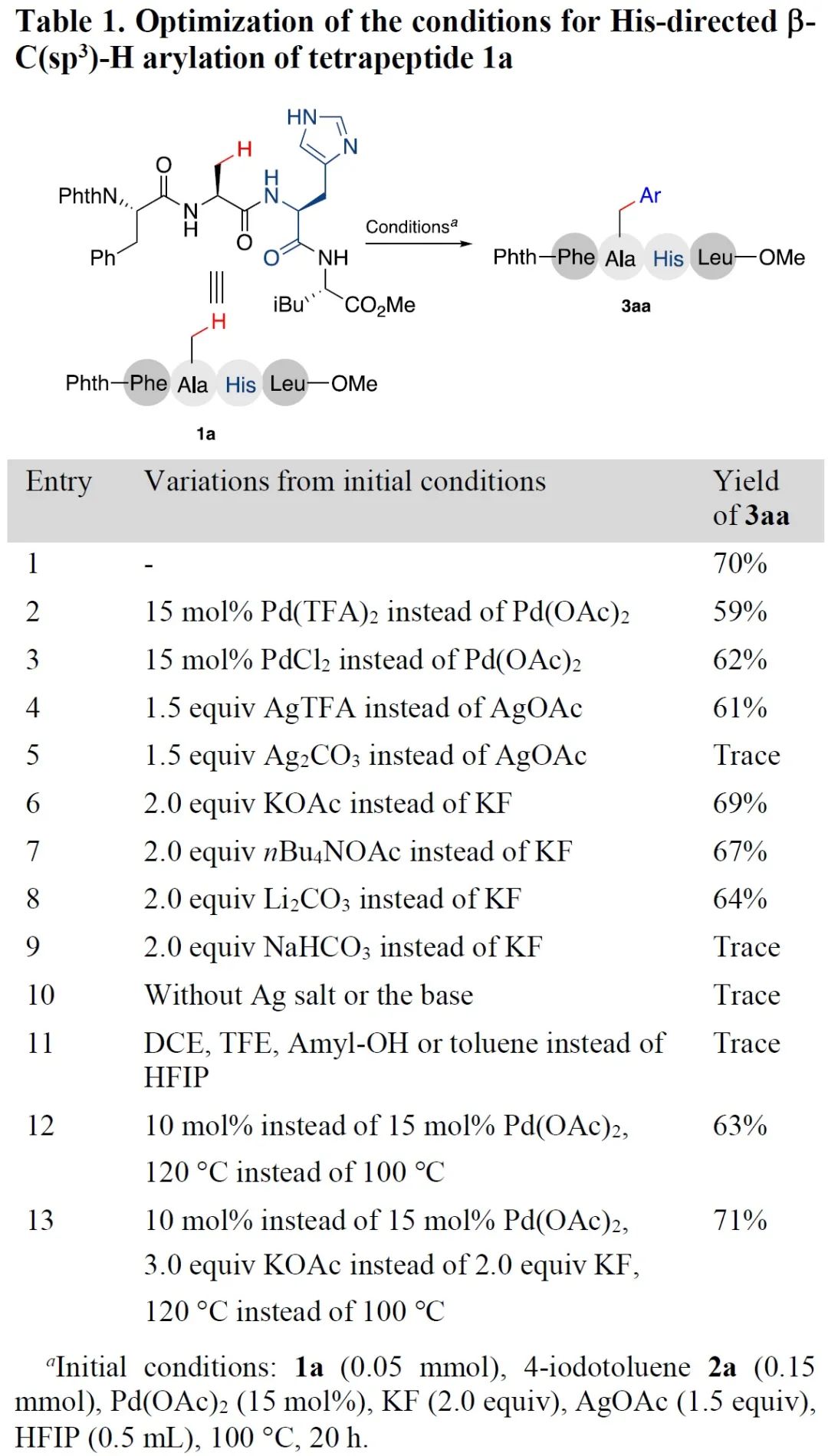
为了验证组氨酸作为导向基团以促进多肽非末端氨基酸残基C(sp3)-H官能团化的可行性,我们以一个含有Ala-His序列的四肽(Phth-Phe-Ala-His-Leu-OMe, 1a)与4-甲基碘苯的反应为模型反应进行研究。经过大量的条件尝试,我们惊喜地发现该反应在15 mol%醋酸钯、1.5当量醋酸银、2当量氟化钾和六氟异丙醇中100°C加热20小时的条件下,以70%的产率得到丙氨酸残基β-C(sp3)-H芳基化产物3aa(Table 1, entry 1)。骨架酰胺N-H和组氨酸侧链咪唑碳氢键芳基化产物均未被检测到。条件优化的同时得到以下重要发现:第一,钯催化剂和银盐的抗衡阴离子对反应结果影响较大。使用阴离子碱性较弱的二氯化钯或三氟乙酸钯代替醋酸钯,以及三氟乙酸银代替醋酸银,均导致产率明显降低(Table 1, entries 2-4)。而使用阴离子碱性较强的碳酸银代替醋酸银则导致反应几乎不发生,仅得到痕量的产物(Table 1, entry 5)。第二,诸如氟化钾、四正丁基醋酸铵、醋酸钾和碳酸锂等弱碱对提高反应的转化率至关重要。在不添加任何碱或使用如碳酸氢钠和碳酸钾等碱的情况下,反应均只得到痕量产物(Table 1, entries 9-10)。第三,溶剂的选择对反应的成功非常关键。在众多溶剂中,只有弱酸性的六氟异丙醇有效(Table 1, entry 11)。其原因可能是六氟异丙醇有利于提高二价钯的亲电性质和二价钯中间体的稳定性。将催化剂的用量进一步降低到10 mol%,再经过一定的条件优化,我们最终确定了最优条件为:1a (0.05 mmol),4-甲基碘苯 2a (3.0 equiv),10 mol% Pd(OAc)2,2.0 当量AgOAc,3.0 当量KOAc,HFIP (0.5 mL),120℃,20小时。在最优条件下,模型反应以71%的产率得到碳氢键芳基化产物。
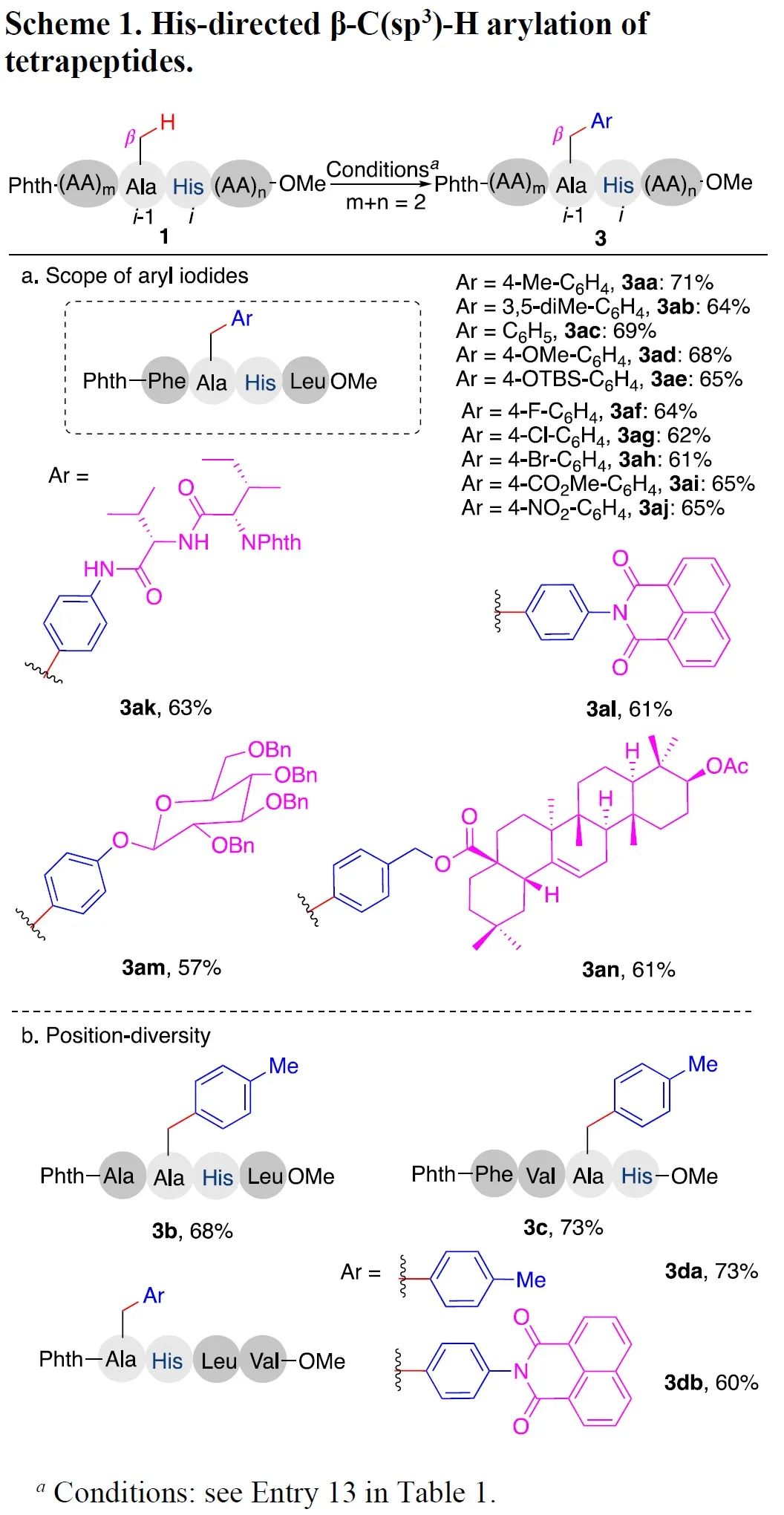
获得最优条件之后,我们考察了组氨酸导向四肽丙氨酸残基β-C(sp3)-H芳基化方法的底物范围。首先,一系列含有吸电子或给电子取代基的芳基化碘化物均与四肽1a反应,以中等以上的产率得到相应的碳氢键芳基化产物(3aa-3aj)。烷基、醚键、硅醚键、卤原子、酯基和硝基等都耐受反应条件。以酰胺键、酰亚胺键、醚键和酯基作为连接子,我们合成了四个连有二肽、荧光分子、D-葡萄糖和齐墩果酸的芳基碘化物,并利用所发展的碳氢键芳基化反应,实现了四肽分子与这些功能分子的高效偶联(Scheme 1a)。除了1a外,其它三个含有Ala-His序列的四肽也以高产率和高位置选择性得到丙氨酸残基β-C(sp3)-H芳基化产物(Scheme 1b)。尤其是含有两个丙氨酸残基的四肽分子1b中,与His相邻的丙氨酸发生了选择性碳氢键芳基化,而氮端丙氨酸则未发生反应。
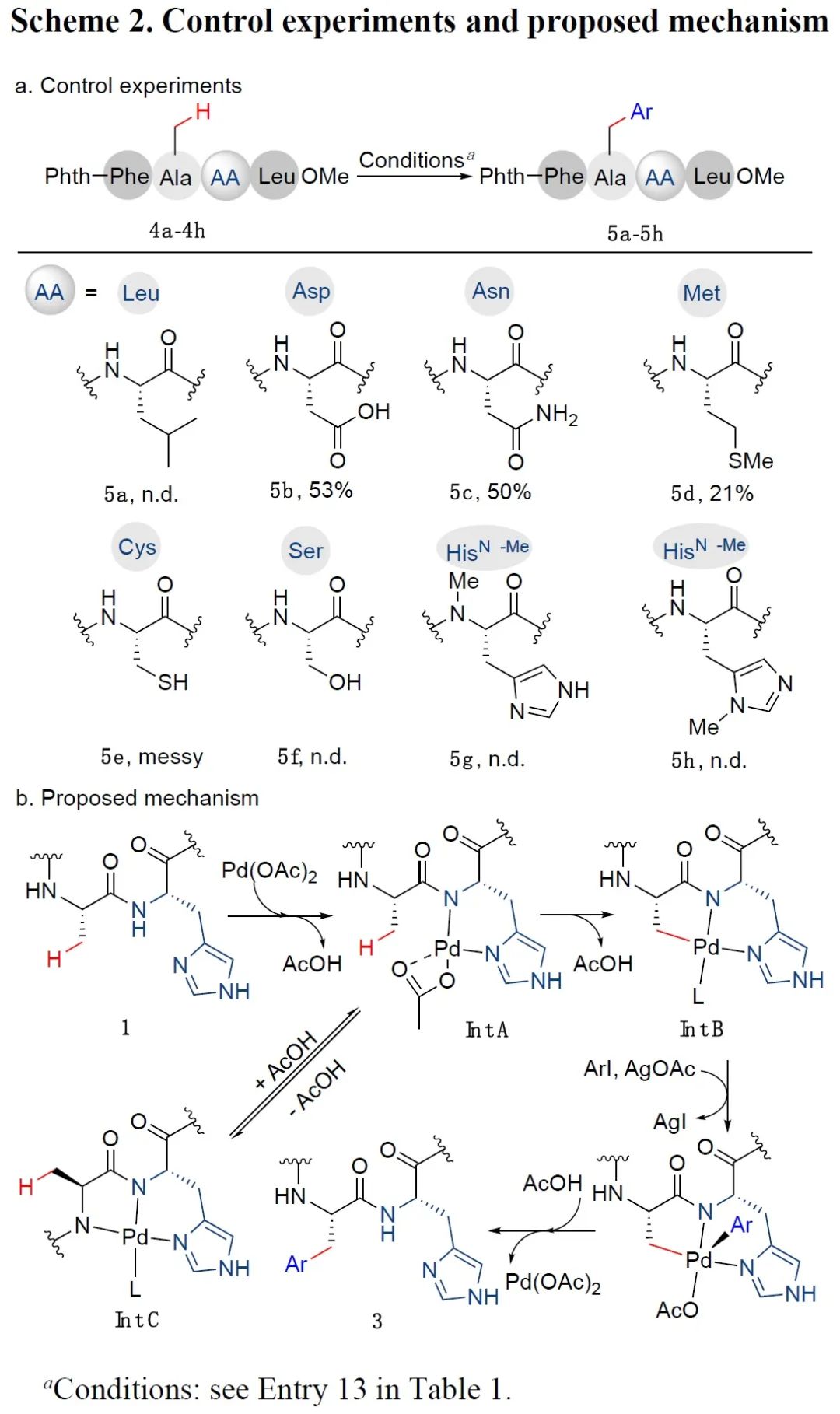
为了探究反应机理,我们开展了一组对比实验(Scheme 2a)。首先,用不含配位性侧链的亮氨酸(Leu)代替1a中的组氨酸(His),碳氢键芳基化反应完全被抑制。之后,我们进一步测试了其它含有配位性侧链的氨基酸的导向能力。含有较强配位性侧链的天冬氨酸(Asp)、天冬酰胺(Asn)或蛋氨酸(Met)替换组氨酸时,所得的四肽底物均发生丙氨酸残基的碳氢键芳基化反应(5b-5d),但是产率低于1a的反应产率。含有较弱配位性侧链的丝氨酸没有表现出导向能力。此外,半胱氨酸因为侧链上巯基的不稳定性,也难以胜任导向基团的角色。以上结果表明侧链的配位能力对于碳氢键芳基化反应至关重要。最后,我们还发现,不管是组氨酸残基的α-氮原子,还是侧链上咪唑氮原子,被选择性甲基化保护之后,碳氢键芳基化均被完全阻断。该结果说明组氨酸残基通过脱去α-N-H的质子之后,作为N,N-双齿导向基团与Pd(II)配位,促进了邻位丙氨酸残基的碳氢键芳基化。基于以上结果,组氨酸导向的碳氢键芳基化反应机理如下(Scheme 2b):首先,多肽1通过组氨酸残基与钯催化剂之间的配位反应,生成多肽- Pd(II)配合物Int A。该配合物先经历协同金属化-脱质子过程,转化为关键中间体Int B,后通过氧化加成和还原消除环节,最终生成碳氢键芳基化产物3。与此同时,Int A也可通过脱质子过程,形成无活性的N^N^N-Pd型配合物Int C。本工作碳氢键芳基化反应成功的原因可能是由于咪唑配体的反位效应削弱了其对位酰胺氮与钯之间的配位键强度从而抑制Int C的生成。
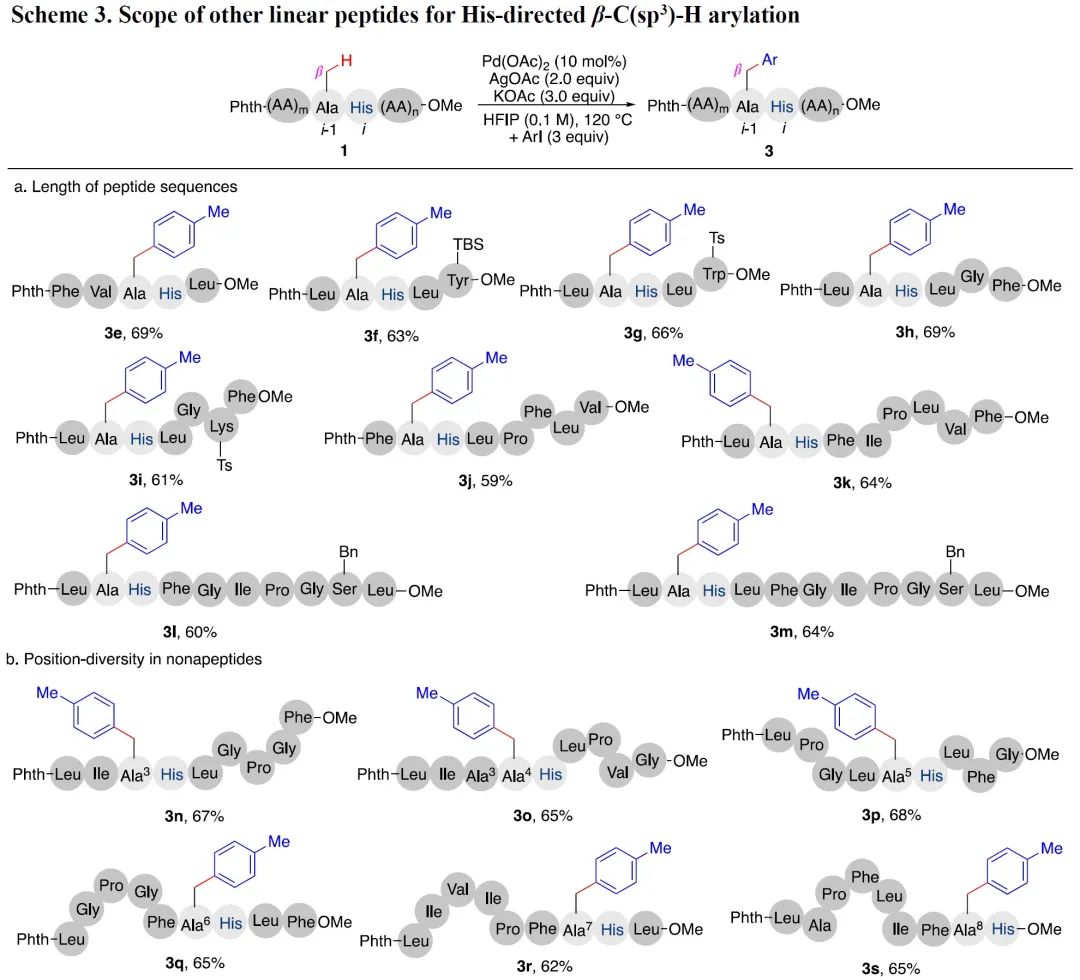
受以上研究结果的激励,我们进一步尝试了更长序列的线性多肽的反应(Scheme 3)。令人高兴的是,随着肽链长度的增加,反应活性没有明显降低。含有Ala-His序列的五、六、七、八、九、十以及十一肽均发生选择性的丙氨酸残基β-C(sp3)-H芳基化反应,以中等偏上的产率得到芳基化产物(3e-3m)。甘氨酸、缬氨酸、亮氨酸、异亮氨酸、脯氨酸、苯丙氨酸、Nπ-Ts色氨酸、Nε-Ts赖氨酸、O-Bn丝氨酸和O-TBS酪氨酸等均很好地耐受反应条件。多肽修饰位置具有多样性,随着Ala-His的位置变化而变化。比如,Ala-His处在氮端+2到+8位的7个九肽分别在+2到+8位发生位置选择性β-C(sp3)-H芳基化(3k, 3n-3s, 62%-68%)。值得一提的是,随着His左侧酰胺键数目的增加,反应活性并没有降低,说明骨架酰胺键对碳氢活化反应的抑制效应得到很好的解决。
综上所述,我们开发了一种钯催化组氨酸导向的β-C(sp3)-H芳基化方法用于多肽的后期修饰。在标准条件下,一系列含有Ala-His序列的线性多肽均发生选择性的丙氨酸残基碳氢键芳基化反应,以中等及以上的产率得到芳基化产物。该修饰方法不仅具有优良的位置选择性,还具有很好的位置多样性。对比实验表明脱质子后的α-氮原子和咪唑氮原子对Pd(II)的螯合作用是反应成功的关键因素。此外,本方法用于多肽与各种功能分子的偶联反应,展示了其在其它领域的潜在应用前景。
